
Qu'est-ce que la maladie de Hirschsprung ?
La maladie de Hirschsprung est une maladie congénitale affectant le tube digestif. Elle est la conséquence d’un défaut de développement du système nerveux dans la paroi du segment terminal de l’intestin.
Sur une personne non malade, des cellules ganglionnaires nerveuses situées dans les plexus sous-muqueux et myentériques assurent le contrôle des muscles lisses de la paroi intestinale. Les plexus se définissent comme des réseaux de nerfs ou de vaisseaux. Le plexus sous-muqueux dit aussi « de Meissner » contrôle les secrétions. Le plexus myentérique dit aussi « d’Auerbach » contrôle quant à lui la motricité.
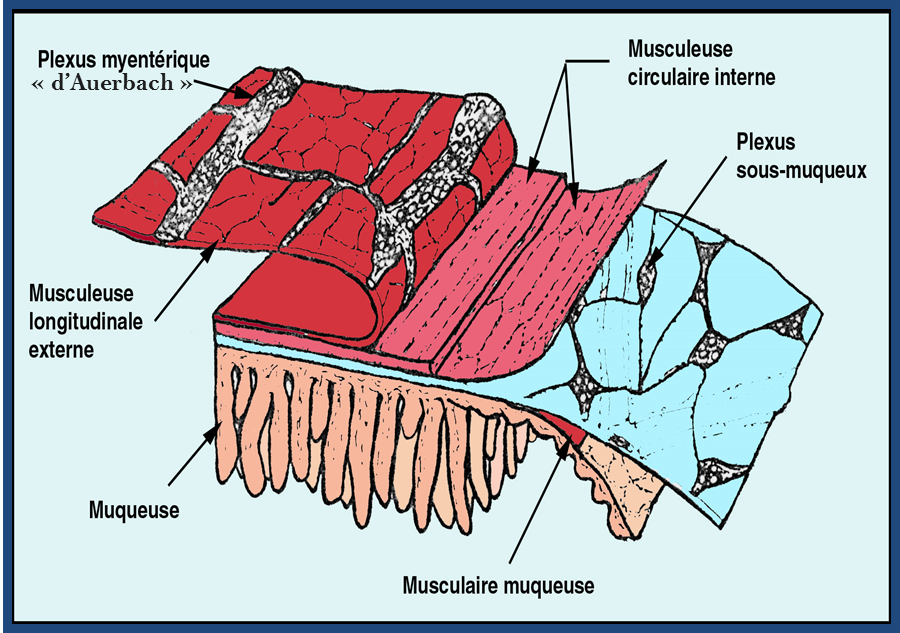
La maladie de Hirschsprung est le résultat de l’absence de cellules ganglionnaires nerveuses dans ces plexus du tube digestif, au niveau de la sous-muqueuse et des muscles de la paroi intestinale. On appelle cette zone d’intestin malade la zone aganglionnaire. Cela entraine une absence de mouvements automatiques de l’intestin permettant la propulsion du bol alimentaire (péristaltisme) et cela peut avoir pour conséquence de créer une occlusion intestinale.
Cette maladie est rare et sa fréquence dans la population est estimée à environ 1 naissance sur 5000. Elle est plus fréquente chez les garçons, qui ont classiquement des formes moins étendues que les filles. Les cas recensés sont majoritairement sporadiques, c’est à dire sans antécédents familiaux. Toutefois, des formes héritées (avec antécédents familiaux) sont rapportées dans 15% des cas. Plusieurs gènes qui prédisposent à l’apparition de la maladie ont été identifiés (on les appelle les gènes de susceptibilité). Le principal gène identifié est le gène RET.
Les formes non syndromiques sont les plus fréquentes, elles sont estimées à 70 % des cas.
Quelle que soit l’origine de la maladie, le segment digestif aganglionnaire se caractérise par une absence de péristaltisme (mouvement automatique de l’intestin) et est à l’origine d’un obstacle fonctionnel digestif.
Sur une personne non malade, des cellules ganglionnaires nerveuses situées dans les plexus sous-muqueux et myentériques assurent le contrôle des muscles lisses de la paroi intestinale. Les plexus se définissent comme des réseaux de nerfs ou de vaisseaux. Le plexus sous-muqueux dit aussi « de Meissner » contrôle les secrétions. Le plexus myentérique dit aussi « d’Auerbach » contrôle quant à lui la motricité.
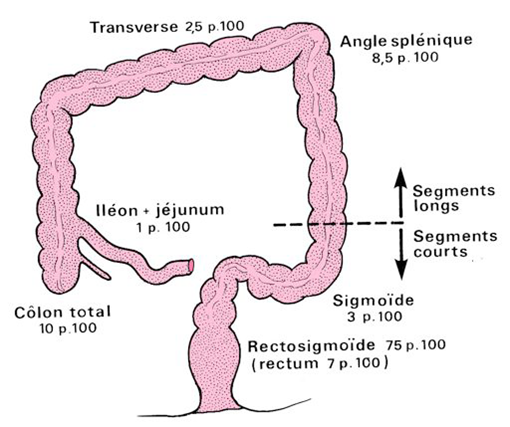
La forme de la maladie la plus fréquente est la forme recto-sigmoïdienne, dit aussi forme courte. Elle concerne 75 % des cas. Elle commence toujours à partir du canal anal et remonte jusqu’au sigmoïde.
Sont également observées des formes longues, c’est à dire qui vont au delà du sigmoïde, sont pancoliques (totalité du colon atteint) ou sont parfois étendues au grêle.
Sur le plan clinique, la maladie est de gravité variable selon la longueur du segment d’intestin atteint et selon les malformations associées s’il s’agit d’une forme syndromique (cardiopathie, épilepsie réfractaire….).
Vie avec la maladie et prise en charge chirurgicale : retrouvez le passage du centre MAREP dans ce reportage du Magazine de la Santé (France 5)
La forme classique se manifeste le plus souvent dès la naissance par des symptômes d’occlusion intestinale. Chez le nouveau-né cela se traduit par un retard d’émission du premier méconium et une occlusion intestinale fonctionnelle avec une dilatation intestinale en amont du segment atteint. Une distension abdominale majeure (augmentation du volume de l’abdomen dû à un ballonnement) sur rétention de méconium, de selles ou de gaz peuvent aussi être les symptômes de la maladie de Hirschsprung. La complication principale est l’entérocolite aiguë.
Des formes atténuées ultra-courtes sont également possibles. Cela concerne moins de 10% des cas. La maladie se manifeste alors par une constipation sévère dont le diagnostic peut n’être posé que plus tard chez le nourrisson ou l’enfant, voire même à l’âge adulte.
Dans certains cas, le diagnostic est plus tardif chez le nourrisson, ou dans de rares cas peu sévères chez le jeune enfant. Une maladie de Hirschsprung peut en effet être révélée par une constipation évoluant depuis la naissance chez un enfant devenu très dépendant des traitements évacuateurs (laxatifs, lavements). Il faut également savoir évoquer ce diagnostic dans un contexte d’entérocolite du nouveau-né.
Sur une personne non malade, des cellules ganglionnaires nerveuses situées dans les plexus sous-muqueux et myentériques assurent le contrôle des muscles lisses de la paroi intestinale. Les plexus se définissent comme des réseaux de nerfs ou de vaisseaux. Le plexus sous-muqueux dit aussi « de Meissner » contrôle les secrétions. Le plexus myentérique dit aussi « d’Auerbach » contrôle quant à lui la motricité.

Dans le cadre du bilan, un lavement opaque aux hydrosolubles est réalisé, pouvant faire suspecter le diagnostic en raison de l'existence d'une disparité de calibre (jonction visible entre le tube digestif sain et la zone atteinte non mobile).
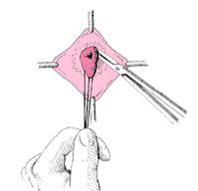
Toutefois, la confirmation du diagnostic doit obligatoirement être faite sur l’étude histologique d’une biopsie rectale superficielle (analyse au microscope d'un petit fragment de paroi rectale prélevé avec la pince de Noblett ou de Scheye au lit du patient). Dans certains cas, la biopsie rectale profonde au bloc opératoire sous anesthésie générale est requise si le doute diagnostic persiste.
Le traitement est chirurgical dans tous les cas et en général réalisé dans les premières semaines de vie.
L’intervention peut consister en une dérivation digestive à la peau (colostomie ou stomie de l’intestin grêle) dans le cas d’une entérocolite et/ou d’une situation digestive instable. Dans tous les cas, seule une suppression du segment d’intestin malade peut traiter le patient. On appelle cela une exérèse. On réalise alors un contrôle histologique peropératoire pour être certain de réaliser la résection (retrait de la partie malade) en zone saine.
Dans les formes coliques totales, la colectomie (retrait du colon) et la résection d’une partie plus ou moins étendue de l’intestin grêle (en fonction de l’atteinte) sont indiquées. D’éminents chirurgiens comme Orvar Swenson (1909-2012 : abaissement colo-anal), Franco Soave (1917-1984 : abaissement extra-muqueux endorectal) et Bernard Duhamel (1917-1996 : abaissement rétrorectal transanal) ont mis au point des techniques encore utilisées aujourd’hui.
L’intervention chirurgicale vise à retirer/couper ou à contourner la zone aganglionnaire et à rétablir la continuité digestive avec le canal anal. Cette intervention a longtemps été réalisée par laparotomie (incision de la paroi abdominale). Aujourd’hui, elle est réalisée le plus souvent en un seul temps opératoire, par voie transanale parfois cœlio-assistée (coelioscopie).
Les associations de patients


![]()